
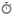
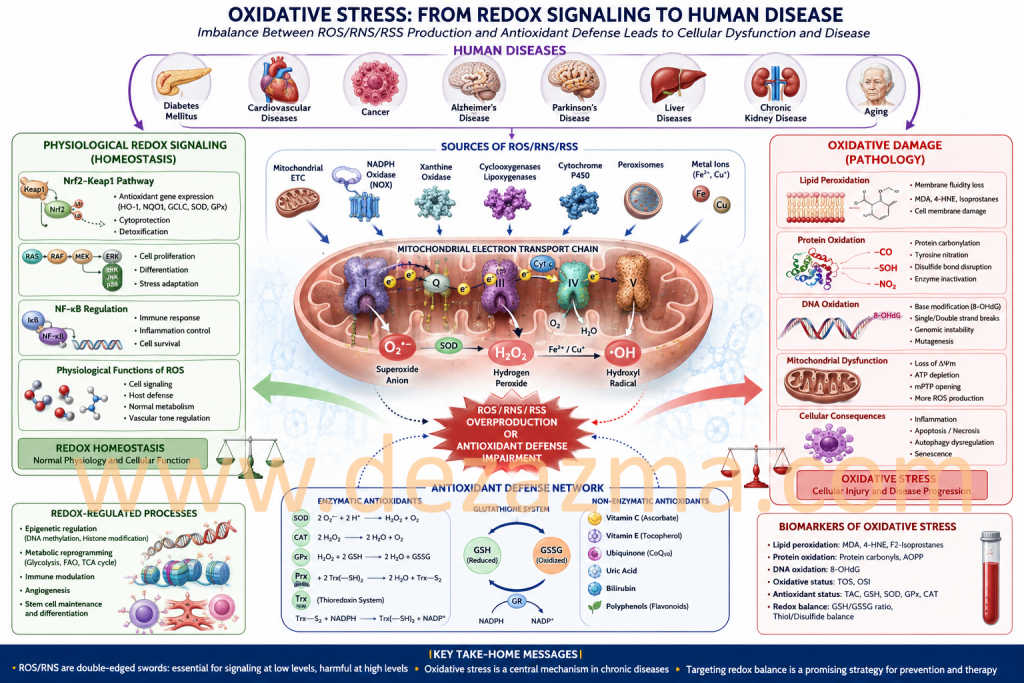
1. Overview of Enzymatic ROS/RNS/RSS Generation
Reactive oxygen, nitrogen, and sulfur species (ROS, RNS, RSS) emerge from a highly integrated enzymatic network distributed across multiple subcellular compartments. Rather than representing stochastic byproducts of metabolism and oxidative stress, these species are generated through specialized redox circuits that function as signaling modules within cellular physiology. Each enzymatic system contributes distinct kinetic and spatial characteristics to intracellular redox dynamics, collectively forming a hierarchical oxidative signaling architecture.¹–³
2. NADPH Oxidase (NOX) Complexes: Dedicated Redox Signal Transducers
NADPH oxidase (NOX) complexes constitute prototypical “professional ROS-generating systems” whose primary biological role is signal transduction rather than energy metabolism. These multi-subunit flavocytochrome assemblies catalyze electron transfer from NADPH to molecular oxygen, producing superoxide (O₂•⁻) as a regulated signaling intermediate.⁴,⁵ Isoform-specific localization (NOX1–5, DUOX1–2) enables compartmentalized ROS generation in the plasma membrane, endosomal membranes, and specialized secretory vesicles.
NOX-derived ROS function as tightly regulated redox bursts that orchestrate immune defense, phagocytic microbial killing, endothelial activation, and growth factor signaling. Importantly, these signals exhibit high spatial fidelity, ensuring that oxidative modifications remain confined to proximal target proteins.⁵,⁶
3. Xanthine Oxidoreductase: Metabolic–Redox Interface Enzyme
Xanthine oxidoreductase (XOR) operates as a bidirectional metabolic–redox switch within purine degradation pathways. Under physiological conditions, XOR predominantly exists as xanthine dehydrogenase; however, proteolytic conversion or oxidative modification shifts enzymatic behavior toward xanthine oxidase, a potent ROS-producing form.⁷
This conversion results in enhanced electron leakage to molecular oxygen, generating superoxide and hydrogen peroxide. XOR-derived ROS become particularly relevant in ischemia–reperfusion injury, where substrate accumulation and oxygen reintroduction converge to produce oxidative bursts that amplify endothelial dysfunction and inflammatory signaling cascades.⁷,⁸
4. Cytochrome P450 Systems: Redox Leakage in Xenobiotic Metabolism
Cytochrome P450 (CYP) monooxygenases represent a vast superfamily of heme-thiolate enzymes localized predominantly in the endoplasmic reticulum. Their canonical role in xenobiotic detoxification and steroid biosynthesis is accompanied by intrinsic electron uncoupling; wherein incomplete reduction of oxygen generates superoxide and hydrogen peroxide as byproducts.⁹
This redox leakage is particularly pronounced under conditions of substrate saturation or metabolic stress, linking hepatic detoxification processes with intracellular oxidative burden. CYP-derived ROS also contribute to lipid peroxidation cascades and electrophilic stress formation.⁹,¹⁰
5. Peroxisomal Oxidases: Compartmentalized Hydrogen Peroxide Production
Peroxisomes function as specialized oxidative organelles enriched in flavin-dependent oxidases that generate hydrogen peroxide as a primary metabolic product. Acyl-CoA oxidase-mediated β-oxidation of very-long-chain fatty acids represents a central source of peroxisomal H₂O₂ production.¹¹
Unlike mitochondrial ATP-coupled oxidation, peroxisomal metabolism decouples electron transfer from energy conservation, resulting in obligatory ROS formation. Catalase and peroxiredoxin systems tightly regulate peroxisomal redox output, preventing cytotoxic accumulation while enabling localized signaling functions.¹¹,¹²
6. Lipoxygenases and Cyclooxygenases: Lipid Radical Chemistry in Inflammatory Signaling
Lipoxygenases (LOX) and cyclooxygenases (COX) catalyze oxygenation of polyunsaturated fatty acids via radical-mediated mechanisms that intrinsically involve reactive oxygen intermediates. These enzymes generate lipid hydroperoxides and prostanoid precursors that serve as bioactive mediators of inflammation, immunity, and vascular tone regulation.¹³
LOX-derived leukotrienes and COX-derived prostaglandins function as both downstream effectors and amplifiers of redox signaling. Their biosynthesis integrates oxidative chemistry with immunological programming, positioning lipid peroxidation pathways as central nodes in inflammation-associated redox networks.¹³,¹⁴
7. Spatial Compartmentalization of Redox Signaling
A defining principle of modern redox biology is the spatial restriction of reactive species generation within subcellular microdomains. Mitochondria, endoplasmic reticulum, peroxisomes, nucleus, and plasma membrane nanoregions each maintain distinct redox microenvironments governed by localized enzymatic production and antioxidant buffering systems.¹,²
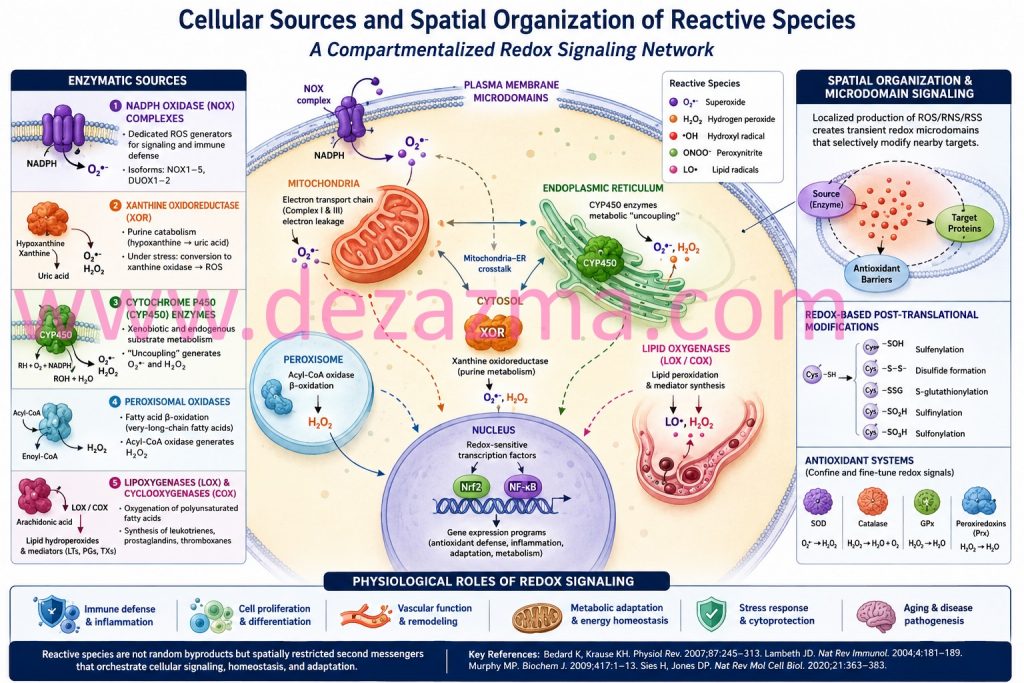
This compartmentalization ensures that ROS act as short-range second messengers rather than diffusible cytotoxins. Within these microdomains, reversible oxidation of cysteine and selenocysteine residues modulates protein conformation, enzymatic activity, and transcriptional output.³
References
Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol. 2015;4:180–183.
Holmström KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signaling. Nat Rev Mol Cell Biol. 2014;15(6):411–421.
Jones DP, Sies H. The redox code. Antioxid Redox Signal. 2015;23(9):734–746.
Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313.
Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4(3):181–189.
Ushio-Fukai M. Compartmentalization of redox signaling through NOX family NADPH oxidases. Free Radic Biol Med. 2009;46(2):129–141.
Pacher P, Nivorozhkin A, Szabó C. Therapeutic effects of xanthine oxidase inhibitors. Pharmacol Rev. 2006;58(1):87–114.
Granger DN, Kvietys PR. Reperfusion injury and reactive oxygen species. J Physiol. 2015;593(2):341–348.
Zangar RC, Davydov DR, Verma S. Mechanisms that regulate cytochrome P450. Toxicol Appl Pharmacol. 2004;199(3):316–331.
Guengerich FP. Cytochrome P450 and chemical toxicology. Chem Res Toxicol. 2008;21(1):70–83.
Wanders RJA, Waterham HR. Biochemistry of mammalian peroxisomes. Annu Rev Biochem. 2006;75:295–332.
Fransen M, Nordgren M, Wang B, Apanasets O. Role of peroxisomes in ROS metabolism. Antioxid Redox Signal. 2012;16(9):1047–1058.
Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol. 2015;15(8):511–523.
Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):986–1000.



{kind=link}